
Alzheimer to choroba zwyrodnieniowa mózgu, która najczęściej powoduje otępienie u osób po 65. roku życia i prowadzi do śmierci. Schorzenie to po raz pierwszy opisał niemiecki neuropatolog Alois Alzheimer i od jego nazwiska pochodzi jej nazwa. Przyczyny Alzheimera właściwie do dziś są niepoznane.
Pierwsza diagnoza choroby Alzheimera
Auguste Deter
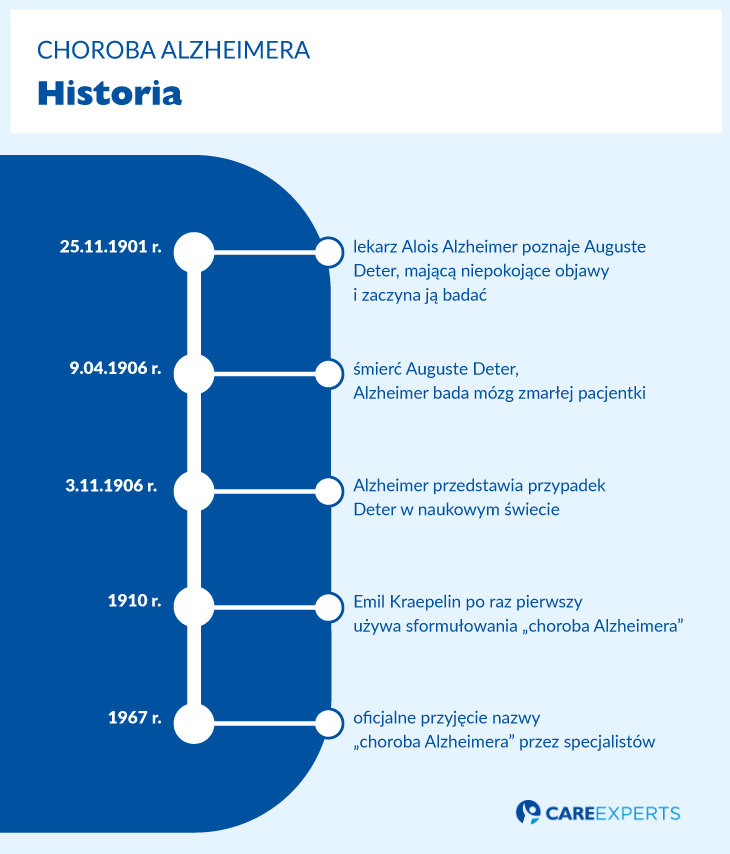
3 listopada 1906 roku Alzheimer przedstawił na posiedzeniu Towarzystwa Neuropsychiatrii Południowo-Zachodnich Niemiec w Tybindze przypadek „specyficznej choroby kory mózgowej” („eine eigenartige Erkrankung der Hirnrinde”) u zmarłej 56-letniej pacjentki, Auguste Deter. Ową pacjentkę, wówczas 51-letnią, poznał 25 listopada 1901 roku w czasie swej pracy w zakładzie psychiatrycznym we Frankfurcie, gdzie przywiózł ją jej mąż. W poprzedzającym roku zauważył w jej zachowaniu znaczne zmiany. Auguste stała się zazdrosna i nie potrafiła wykonywać najprostszych czynności domowych. Chowała przedmioty tak, że nie potrafiła ich znaleźć. Czuła się śledzona i naprzykrzała się sąsiadom.
Alzheimer stwierdził, że pacjentka nie posiada orientacji czasoprzestrzennej. Nie potrafi też przypomnieć sobie prawie żadnych szczegółów z życia i często daje odpowiedzi niemające żadnego związku z pytaniem. Ponadto jej nastroje szybko się zmieniały pomiędzy lękiem, nieufnością, odrzucaniem i popłakiwaniem. Nie można było pozwolić jej na swobodne poruszanie się po zakładzie, bo miała zwyczaj dotykać twarzy innych pacjentów, na co ci często reagowali agresją.
Choroba zapominania
Wprawdzie Alzheimer już wielokrotnie wcześniej natykał się na pacjentów z podobnymi objawami, byli oni jednak w wieku przeważnie co najmniej 70 lat. Alzheimer poświęcił pacjentce wiele uwagi, cierpliwie prowadząc przez najbliższe tygodnie dalsze rozmowy. Podczas tych rozmów Auguste wielokrotnie żałośnie powtarzała „ach Gott” (o Boże). Podczas jednej z rozmów kilkakrotnie wyznała „można by powiedzieć, że sama się zagubiłam” (ich habe mich sozusagen selbst verloren). Alzheimer już po kilku miesiącach przestał pracować we Frankfurcie, ale z Heidelbergu i później z Monachium regularnie dowiadywał się o stan jej zdrowia. Zapobiegł też planowanemu przeniesieniu jej do tańszego zakładu, ponieważ zależało mu na możliwości zbadania mózgu pacjentki po jej śmierci.
Auguste Deter zmarła 9 kwietnia 1906 w wyniku zakażenia krwi, do którego doszło w ranach spowodowanych odleżynami. Na prośbę Alzheimera przesłano mu do Monachium jej mózg i historię choroby. Badanie mikroskopowe wykazało rozległe zwyrodnienie komórek nerwowych. Alzheimer określał przedstawianą przez siebie chorobę jako choroba zapominania („Krankheit des Vergessens”). Określenia „choroba Alzheimera” użył po raz pierwszy Emil Kraepelin w swoim Podręczniku psychiatrii z 1910 roku. Oficjalnie nazwa ta została przyjęta na kongresie lekarzy w Lozannie w 1967 roku.
Alzheimer przyczyny – co dziś wiemy?
O ile objawy choroby Alzheimera – zwłaszcza na późniejszym etapie choroby – są dość jasne do określenia – przyczyna choroby Alzheimera pozostaje zasadniczo nieznana, pomijając od 1 do 5% przypadków, w których zidentyfikowano odmienności genetyczne, przyczynę próbuje wyjaśnić kilka konkurencyjnych hipotez. Obecnie na chorobę Alzheimera choruje na świecie blisko 35 mln ludzi, w Polsce, według Alzheimer Europe, jest to blisko 300 000 osób. Uważa się, że zachorowalność na chorobę Alzheimera podwaja się co 5 lat u osób pomiędzy 65 a 85 rokiem życia. Po ukończeniu 85 lat liczba przypadków rośnie do 40%. Wzrost liczby chorych z chorobą AD będzie dotyczył przede wszystkim krajów o średnim i niskim dochodzie narodowym.
Potwierdzono, że choroba Alzheimera charakteryzuje się długim, blisko 20-letnim okresem przebiegu niemego klinicznie. Dzięki plastyczności mózgu proces patologiczny niszczący tkanki mózgu trwa bardzo długo, zanim chory zaczyna zgłaszać pierwsze problemy, najczęściej dotyczące zaburzeń pamięci.
Hipoteza amyloidowa jako próba określenia przyczyny choroby Alzheimera
Przyczyną obumierania neuronów w chorobie Alzheimera jest odkładanie się w mózgu toksycznych białek o strukturze beta- fałdowej, które powodują największe uszkodzenia w postaci oligomerycznej. To właśnie oligomery wyzwalają procesy apoptozy, która jest naturalnym procesem zaprogramowanej śmierci komórki, oraz martwicy neuronów. Beta- amyloid (Aβ) jest 43- 44 aminokwasowym fragmentem dłuższego peptydu, stanowiącego jego prekursor. Gen białka prekursorowego zlokalizowany jest na chromosomie 21. Znanych jest już kilkadziesiąt mutacji sprawczych w obrębie tego genu.
Prawidłowa degradacja prekursora beta- amyloidu przebiega wtedy, kiedy działa enzym o nazwie alfa- sekretaza. Jeżeli natomiast cięcie zostanie dokonane przez enzymy patologiczne: beta- sekretazę i potem gamma- sekretazę uwalnia się beta- peptyd, najpierw w postaci oligomerów, potem zaś zaczyna agregować w mózgu, tworząc blaszki starcze. Gamma- sekretaza – drugi patologiczny enzym biorący udział w patologicznej degradacji prekursora, wchodzi w skład kompleksu presenilin. Są to: presenilina 1, kodowana na chromosomie 14, lub presenilina 2, kodowana na chromosomie 1.
Mutacje genów dla presenilin (blisko 200), tak jak i te w genie dla prekursora, charakteryzują się blisko 100% penetracją, powodują agresywną, szybko postępującą formę choroby Alzheimera o wczesnym początku (pomiędzy 55 a 60 rokiem życia), dziedziczoną w sposób autosomalnie dominujący. Pamiętać należy jednak, że liczba przypadków o znanym jednogenowym dziedziczeniu dotyczy nie więcej niż 1,5% wszystkich przypadków choroby Alzheimera.
Istotnym zjawiskiem jest także fakt, że obecność wszystkich patologicznie ustrukturyzowanych białek powoduje aktywację komórek glejowych w mózgu, zarówno astrogleju, jak i mikrogleju, co uruchamia procesy towarzyszące zapaleniu: uwalnia wolne rodniki, aminokwasy pobudzające, zapalne interleukiny oraz tlenek azotu. Stanowią one dodatkową przyczynę obumierania neuronów i ich połączeń. Zanik neuronów w oczywisty sposób prowadzi do wypadania ich funkcji, a tym samym spadają stężenia produkowanych przez nie neuroprzekaźników.
W przypadku choroby Alzheimera kluczowym neuroprzekaźnikiem jest acetylocholina. Bierze ona aktywny udział w procesie zapamiętywania, a także inne przekaźniki, np. te odpowiadające za nastrój, takie jak serotonina i noradrenalina.
Badanie przyczyn Alzheimera – hipoteza cholinergiczna
Najstarsza jest hipoteza cholinergiczna, na której bazują dostępne obecnie sposoby farmakoterapii. Według niej za chorobą Alzheimera stoi zmniejszona synteza neuroprzekaźnika acetylocholiny. Zaburzone funkcjonowania układu cholinergicznego, którego podstawowe elementy stanowią: acetylotransferaza cholinowa (ang. choline actyltransferase – ChAT), acetylocholinoesteraza (acetylcholinesterase – AChE), precyzyjny wychwyt choliny (ang. high-affinity choline uptake – HACU) i receptory cholinergiczne: muskarynowe (muscarinic receptors – mAChR) i nikotynowe (nicotinic receptors – nAChR).
Na podstawie licznych badań stwierdzono znaczącą i wybiórczą utratę aktywności ChAT w różnych częściach mózgu u pacjentów z chorobą Alzheimera (kora, hipokamp, jądro migdałowate) oraz degenerację neuronów w jądrze podstawnym Meynerta. Ponadto zaobserwowano także zmniejszenie wychwytu choliny i uwalniania acetylocholiny, neuroprzekaźnika odgrywającego główną rolę w procesach pamięciowych i uczenia się.
Hipoteza ta zyskała na znaczeniu, ponieważ współczesna terapia opiera się jedynie na lekach będących inhibitorami cholinoesteraz, enzymów odpowiedzialnych za rozkład acetylocholiny.
Cholinoesterazy
Cholinoesterazy to hydrolazy estrów karboksylowych (EC 3.1.1). Bez wątpienia najważniejszym ich zadaniem jest hydroliza acetylocholiny w połączeniach nerwowo-mięśniowych, przez co inaktywują neuroprzekaźnictwo cholinergiczne. W obrębie rodziny cholinoesteraz wydzielono acetylocholinoesterazę AChE (EC 3.1.1.7) i butyrylocholinoesterazę BChE (EC 3.1.1.8; niespecyficzna cholinoesteraza, ChEII, pseudocholinoesteraza, cholinoesteraza osoczowa). Cholinoesterazy są w większości enzymami zewnątrzkomórkowymi. Występują w postaci rozpuszczalnej albo są przymocowane do zewnętrznych powierzchni komórek. U człowieka całkowity poziom cholinoesteraz i dystrybucja ich molekularnych form różni się między poszczególnymi regionami mózgu.
Funkcje cholinoesteraz
AChE znajduje się głównie w neuronach, a BChE związana jest przede wszystkim z komórkami glejowymi. Obydwa enzymy występują w ośrodkowym układzie nerwowym. AChE znaleziono w cholinergicznych synapsach ośrodkowego układu nerwowego, a także poza nim, oraz w nerwach niecholinergicznych. W związku z pełnionymi przez nią funkcjami zlokalizowana została w erytrocytach, trombocytach i płytkach krwi. Prawdopodobnie występuje również w rdzeniu nadnerczy.
BChE jest z kolei syntetyzowana w wątrobie, a następnie wydzielana do osocza. Razem z AChE zostały znalezione w przewodzie pokarmowym i w sercu. Najlepiej poznaną funkcją cholinesteraz jest hydroliza acetylocholiny, a przez to terminacja przekaźnictwa cholinergicznego. Poza estrami choliny BChE jest zdolna do rozkładu innych estrów oraz związków toksycznych, jak na przykład: pestycydy, kokaina, octany, heroina czy aspiryna. W ten sposób chroni ona organizm człowieka przed zatruciem. Istnieją liczne badania sugerujące, że cholinoesterazy mogą pełnić rolę peptydaz i amidaz.
Poza aktywnością hydrolityczną AChE i BChE pełnią także inne funkcje. Uczestniczą w neurogenezie i synaptogenezie, wykazują aktywność neurotroficzną i uczestniczą w proliferacji, różnicowaniu i adhezji komórek. Biorą także udział w przekazywaniu sygnałów, regulowaniu bariery krew-mózg, metabolizmie energetycznym, reakcji na stres i aktywności motorycznej. Odkryto również ekspresję AChE w fotoreceptorach. Enzym ten jest także zaangażowany w różnicowanie się komórek krwi. Istnieją doniesienia o nienormalnej aktywności cholinoesteraz w niektórych typach komórek nowotworowych.
Cholinoesterazy w chorobie Alzheimera
W mózgach pacjentów z zaawansowaną chorobą Alzheimera stwierdzono znaczny wyrost (41-80%) aktywności BChE, podczas gdy aktywność AChE pozostaje niezmieniona lub ulega obniżeniu. Dla porównania w zdrowym mózgu za regulację poziomu acetylocholiny w większym stopniu – 80% odpowiada AChE, natomiast BChE za pozostałe 20%. Deficyty w układzie cholinergicznym odzwierciedla 10-60% utrata AChE w regionach mózgu dotkniętych przez chorobę Alzheimera (kora nowa, hipokamp). Ubytek ten dotyczy szczególnie jej molekularnej formy G4 w płacie ciemieniowym.
Obniżenie aktywności enzymatycznej w mózgu
Liczne badania potwierdziły obniżenie aktywności enzymatycznej i stosunku formy G4/G1 w różnych polach Brodmanna na przyśrodkowej powierzchni półkuli mózgu i w jądrze migdałowatym, przy jednoczesnym powolnym, ale znaczącym wzroście form G1 i/lub G2 obydwu cholinoesteraz. U zdrowego człowieka uznano bowiem, że w ośrodkowym układzie nerwowym fizjologiczne funkcje cholinoesteraz spełnia przede wszystkim forma G4 AChE (70-75% aktywności), a w mniejszym stopniu G1.
Na tej podstawie zaproponowano przeważającą stratę formy G4 w chorobie Alzheimera, która powiązana jest z degeneracją elementów presynaptycznych zaangażowanych w regulację transmisji AChE, przy jednoczesnym zaoszczędzeniu korowych form G1. Jest ona prawdopodobnie związana ze strukturami postsynaptycznymi, nie dotkniętymi zmianami chorobowymi. Natomiast stosunek G4/G1 dla BChE ulega obniżeniu o 20-40%. U chorych na Alzheimera zauważono 20% wzrost aktywności formy G1 BChE, podczas gdy forma G4 pozostaje bez zmian.
Zmiana w aktywności cholinoesteraz BChE/AChE
W chorobie Alzheimera, zależnie od lokalizacji w mózgu, następuje również zmiana stosunku BChE/AChE. I tak w płacie czołowym odnotowano wzrost tej proporcji z 0,6 do 0,9, a w korze entorinalnej z 0,6 do 11. Dokładniejsze analizy zmian w dystrybucji form AChE w 21. polu Brodmanna u chorych ze zdiagnozowaną chorobą Alzheimera wykazały, że nie ma zmian w rozkładzie rozpuszczalnych form G1 i G4, przy jednocześnie znaczącym (40%) spadku związanych z błonami form G4 AChE. Ponadto zauważono wzrost aktywności asymetrycznych form A12 i A8, odpowiednio o 342% i 406%. W zdrowym mózgu występują one w niewielkich ilościach. Podwyższony poziom form asymetrycznych jest łączony ze zmianami patologicznymi w chorobie Alzheimera.
Na początku lat 90. XX stulecia przeprowadzono analizy nagromadzenia aktywności AChE w częściach mózgu, pochodzącego od chorego na Alzheimera, w których stwierdzono duże ilości płytek starczych i zwłóknień neurofibrylarnych. Otrzymane wyniki pozwoliły przypuszczać, że w tych obszarach mogą występować formy asymetryczne, zakotwiczone w nich za pomocą kolagenowego ogona. Na podstawie kolejnych badań zwrócono uwagę, że poza rolą w układzie cholinoergicznym AChE może przyspieszać formowanie amyloidu β. Jest to zgodne z poglądem, że zarówno rozpuszczalna, jak i zakotwiczona w błonie AChE, sprzyja tworzeniu fibryli amyloidu w warunkach in vivo. Powstające kompleksy AChE i amyloidu β są bardziej toksyczne niż sam amyloid β, przy czym kompleks AChE z Aβ 1-40 jest bardziej toksyczny niż z Aβ 1-42. Natomiast odpowiedź na pytanie o rolę BChE w tworzeniu złogów amyloidowych jest niejednoznaczna.
Według niektórych badaczy BChE jest powiązana z płytkami neurytycznymi odkładającymi się w mózgach pacjentów ze zdiagnozowaną chorobą Alzheimera. Zaobserwowano jej akumulację w blaszkach formowanych w późniejszych stadiach chorobowych. Jednak w dalszym ciągu pozostaje dyskusyjnym fakt, czy BChE uczestniczy w dojrzewaniu tych zmian neuropatologicznych. Istnieją także wyniki badań sugerujące, że poza tworzeniem kompleksów z amyloidem, obie cholinoesterazy tworzą także kompleksy z białkiem tau.
Hipoteza białka tau
Prowadzone w latach osiemdziesiątych badania nad chorobą Alzheimera pozwoliły stwierdzić, że w mózgu chorych, poza złogami blaszek starczych zbudowanych głównie z β -amyloidu, występują również inne zwyrodnienia znajdujące się wewnątrz komórek nerwowych i nazywane splotami włókienek nerwowych (NT).
Zwyrodnienia te przypominają wielokrotnie skręcone dwie, splecione ze sobą nici, tworzące spiralę i zbudowane są z wysoko ufosforylowanego białka tau. Wykazano, że w mózgu pacjentów z chorobą Alzheimera nadekspresja białka tau i wzrost poziomu kinaz MARK powoduje odłączenie tau od mikrotubul i tworzenie PHF. Jest to przyczyną uszkodzenia transportu organelli do zakończeń aksonalnych i synaps, przede wszystkim mitochondriów, co prowadzi do poważnych zaburzeń równowagi energetycznej neuronów.
Z wiekiem uszkodzeniu ulega cytoszkielet komórkowy i tym samym zaburza transport aksonalny, co również prowadzi do śmierci neuronów. Znaczącą rolę w powstawaniu patologicznych złogów cytoszkieletu odgrywa białko tau. Wskutek zaburzenia równowagi pomiędzy procesami fosforylacji i defosforylacji wzrasta ilość wysoce ufosforylowanych izoform tego białka niezdolnych do stabilizacji aparatu mikrotubularnego neuronów.
Wzmocnienie patologicznych przemian białka tau
Najnowsze dane doświadczalne wskazują, że istnieje związek pomiędzy patologiczną przemianą białka APP a zaburzeniami w strukturze cytoszkieletu i tworzeniem się splotów włókienek nerwowych. Uważa się, że skutkiem mutacji w genie kodującym presenilinę 1 jest zmiana jej właściwości, co powoduje modulację procesu fosforylacji białka włókienkowego NF-H (ang. neurofilam ent-H). Białko to reguluje z kolei proces fosforylacji białka tau. Nadmierna fosforylacja tau zmienia jego powinowactwo do mikrotubul i prowadzi do ich destabilizacji. Skutkiem tego jest zaburzenie transportu aksonalnego rozlicznych białek, w tym także białka APP. Powoduje to nadmierną akumulację białka APP i jego nieprawidłową degradację. Patologiczne przemiany białka tau ulegają w tym cyklu wzmocnieniu. Wykazano bowiem, że zarówno białko APP, jak i β- amyloid nasilają lub wręcz indukują proces hiperfosforylacji białka tau.
Według badań przeprowadzonych przez Georgetown Ubniversity Medical Center obumieranie neuronów zachodzi, kiedy tau nie spełnia swojej funkcji. Polega ona na zapewnieniu struktury, która umożliwia komórce usunięcie niechcianych bądź toksycznych białek. Kiedy tau jest nieprawidłowe, białka te, w tym beta-amyloid, akumulują się w neuronach. Komórki zaczynają je wyprowadzać do przestrzeni pozakomórkowej (neuropilu), tak by nie mogły działać toksycznie w ich wnętrzu.
Starzenie i mutacja białka tau
Ponieważ beta-amyloid jest lepki, zbija się w płytki. Badania sugerują, że komórki niszczy beta-amyloid, którego nie udało się wypchnąć. Gdy tau nie działa prawidłowo, neurony nie mogą się pozbywać odpadów, na które składają się zarówno Aβ, jak i zmiany neurofibrylarne (anormalne tau). Neurony obumierają, a uwalniany z nich beta-amyloid przywiera do tworzącej się blaszki. Eksperymenty na modelach zwierzęcych pokazały, że przy działającym tau formuje się mniej blaszek. Kiedy białko tau wprowadzano do neuronów, które go nie miały, blaszki nie rosły. Nieprawidłowe tau jest skutkiem mutacji oraz starzenia. W przebiegu tego ostatniego część białka może nie działać prawidłowo, lecz jeśli prawidłowo zbudowanej reszty jest wystarczająco dużo, neurony radzą sobie z usuwaniem odpadów i nie obumierają. To wyjaśnia dezorientujące obserwacje kliniczne starszych osób, u których występują blaszki, ale nie demencja.
Czynniki środowiskowe
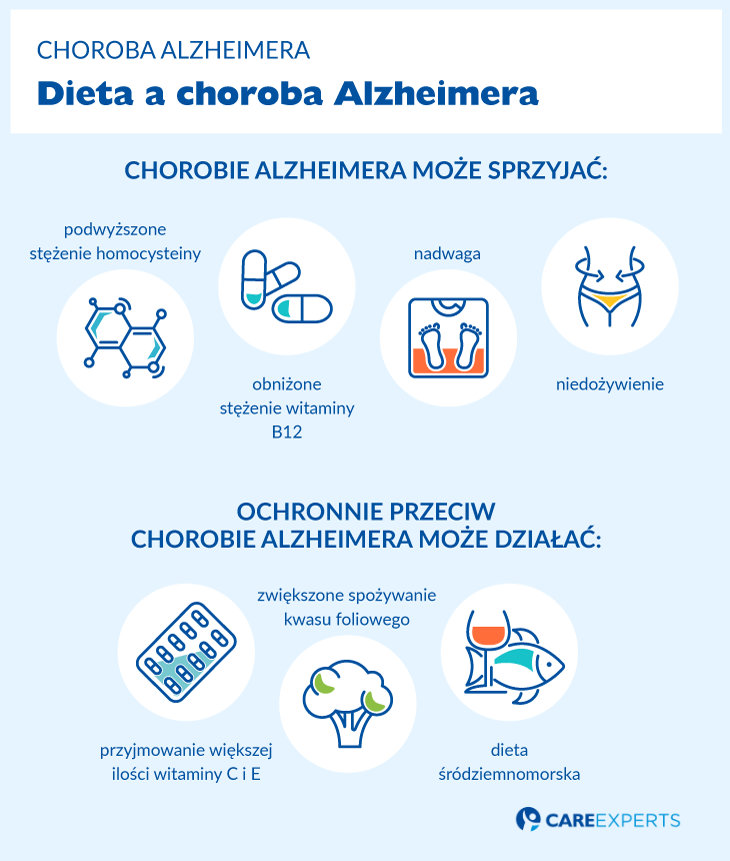
Uważa się, że sposób odżywiania zbliżony w składzie do diety śródziemnomorskiej ma ochronne działanie i sprzyja długotrwałemu życiu w zdrowiu. Podwyższone stężenie homocysteiny oraz obniżone stężenie witaminy B12 może pośrednio wpływać na rozwój choroby Alzheimera. Natomiast zwiększone spożycie kwasu foliowego, witaminy C i E może działać ochronnie.
Niekorzystny wpływ na rozwój AD odnotowano zarówno w przypadku zbyt dużej masy ciała, jak i w przebiegu niedożywienia. Podkreśla się również związek pomiędzy naczyniopochodnym uszkodzeniem mózgu a rozwojem otępienia.
Najważniejszym czynnikiem środowiskowym który przyspiesza zwyrodnienia jest stan naczyń mózgowych. Przemawia za tym fakt, że odpowiednia dieta, prawidłowy stan odżywienia, brak cukrzycy, unikanie palenia papierosów oraz zwrócenie uwagi na aktywność fizyczną odgrywają istotną rolę w zapobieganiu nie tylko chorobom układu sercowo-naczyniowego, ale także AD.
Dane epidemiologiczne dowodzą, iż wraz ze starzeniem się społeczeństwa częstość występowania AD będzie wzrastać, tym samym zwiększy się liczba osób niezdolnych do samodzielnego funkcjonowania.
Opracowanie: Paulina Mikołajczyk, psycholog Care Experts
Opieka nad osobami chorymi na Alzheimera
|
Zobacz podobne artykuły:
Alzheimer – objawy i stadia choroby
Alzheimer – etapy choroby
Alzheimer – leczenie i opieka
Choroba Alzheimera – test
Alzheimer jest dziedziczny?
Choroba Alzheimera – gdzie szukać pomocy?
Bliski ma Alzheimera – jak się przygotować na przyszłość?
Opieka nad chorym na Alzheimera – komunikacja i aktywności
Literatura tematu:
Choroba Alzheimera: obecny stan wiedzy, perspektywy terapeutyczne, Jerzy Leszek, https://journals.viamedica.pl/polski_przeglad_neurologiczny/article/
Choroby neurodegeneracyjne: choroba Alzheimera i Parkinsona, Małgorzata Gaweł, Anna Potulska-Chromik, http://www.pnmedycznych.pl/wp-content/uploads/2015/07
Rola nauk biologicznych w zrozumieniu genezy i nowego podejścia terapeutycznego do choroby Alzheimera, E Tęgowska, A. Wosińska, http://www.phmd.pl/api/files/view/26112.pdf
Molekularne podłoże Choroby Alzheimera, http://www.e-biotechnologia.pl/Artykuly/alzheimer-molekularne-podloze
Budowa i rola białka tau, Marta Baksalerska-Pazera, Grażyna Niewiadomska, www.pb.ptbioch.edu.pl/pdf/4_2002/287
Komentarze zostały wyłączone.